Projects
Spinal Mechanisms Underlying SCI-Induced Pain: Implications for Targeted Therapy
SCI causes long-term disability including loss of motor and sensory function, as well as chronic neuropathic pain in up to 80% of patients that respond poorly to conventional analgesic therapies. We have been examining the role of specific CCA pathways not only in secondary spinal cord tissue damage after SCI but also as a critical mechanism for the development of central neuropathic pain. A highly synergistic collaboration between the Faden and Dorsey laboratories has linked the trkB.T1 receptor to posttraumatic hyperalgesia after SCI through regulation of cell cycle pathways.
Based on transgenic model work, we identified a promising new therapeutic target: a truncated isoform of the brain-derived neurotrophic factor (BDNF) receptor—tropomyosin-related kinase B (trkB), or trkB.T1 (Wu et al., 2013). Although BDNF signaling through its full-length receptor isoform (trkB.FL) is pro-nociceptive, our studies point to an anti-nociceptive phenotype when trkB.T1 is deleted in vivo. However, the precise cellular mechanisms underlying this finding are not fully understood. The purpose of this study is to investigate how trkB.T1 drives post-injury neuropathic pain via astrocyte dysfunction and test the hypothesis that astrocytic trkB.T1 functions as a key mechanism in post-injury reactive astrogliosis, through altered transcriptional programming that controls cellular movement and immune function, thus affecting chronic pain after SCI.
We examined if trkB.T1 signaling in astrocytes alters transcriptional programming that controls cellular movement and immune function. The migratory and proliferative capacities of astrocytes in the presence or absence of TrkB.T1 were analyzed using the xCELLigence real-time cell analysis system. We showed that KO astrocytes exhibited slower migration/proliferation in vitro in response to 10% FBS or 50 ng/ml BDNF compared with WT astrocytes (Matyas et al., 2017). To assess astrocyte activation status after SCI, ACSA-2+ astrocytes were isolated from moderate injured spinal cord tissue at sham and 7d after injury, as reactive astrocytes begin to form glial scar at 7 days post-injury. The RNA seq analysis revealed downregulation of cell proliferation pathways in KO astrocytes, consistent with the data derived from cultured astrocytes. In addition, there were robust differential changes between TrkB.T1 KO and WT in Cytokine-cytokine receptor interaction pathway, antigen processing pathway, and defense response pathway. These observations may lead to novel therapeutic targets for neuropathic pain in a wide range of disease states.
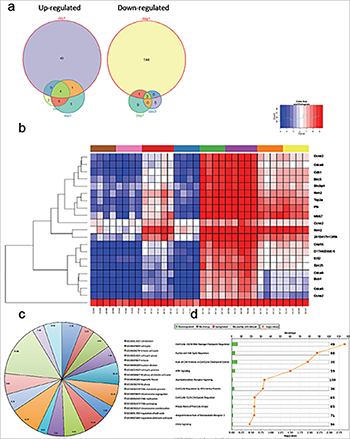
Figure 1. Cell cycle genes are upregulated in the spinal cord of trkB.T1+/+ mice early after SCI but not in the trkB.T1 null. Differential gene expression analysis over time demonstrates that the largest number of genes is altered early after SCI, at day one. The Venn diagrams show each time point, with the number of differentially expressed genes. Heat map of genes that cluster by genotype and time in trkB.T1-/- spinal cord compared with wildtype. The normalized expression of the summarized probe sets is shown. The histogram key indicates that blue genes are down-regulated and red genes are up-regulated. The pie chart lists the 17 significantly enriched GO terms in knockout versus wildtype spinal cord at day one. The numbers in each slice represent the degree of enrichment; all are statistically significant at FDR P < 0.02. Unbiased pathway analysis demonstrates significant down-regulation of cell cycle pathways in trkB.T1-/- spinal cord compared with wildtype. All are significant per the –log10 p values shown as orange squares (n=4-7).
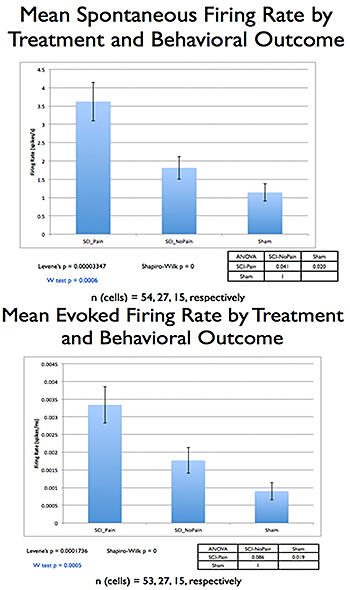
Figure 2. Comparing SCI rats with or without post-injury hyperalgesia, and sham-operated controls, showed significant differences (p=0.005, Kruskal-Wallis ANOVA), with a significant correlation between increases in the posterior thalamic nucleus (PO) spontaneous/evoked activity and hyperalgesia.